
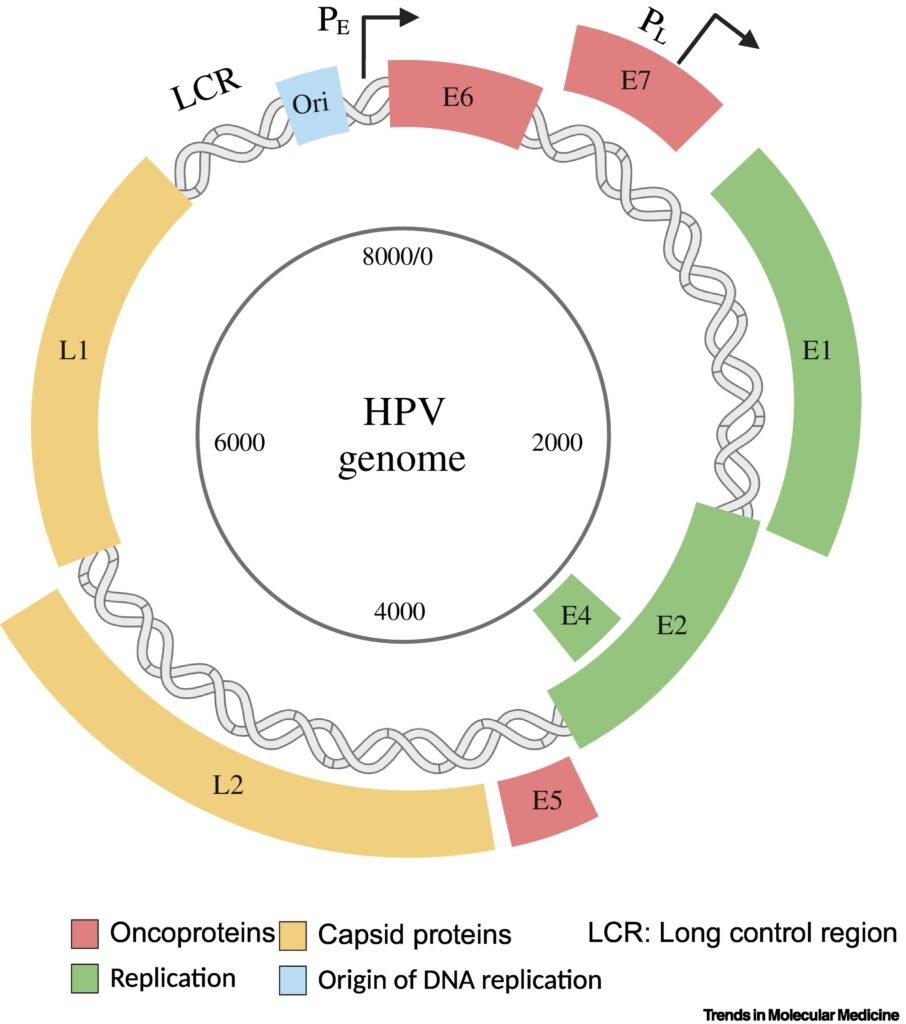
HPV infection is considered the main driver of oncogenesis in the cervix. HPV is a non-enveloped DNA virus with a double-stranded genome containing ~8000 base pairs . More than 400 HPV genotypes in the Papillomaviridae family have been identified to date, which have been categorized into the five genera Alpha, Beta, Gamma, Mu, and Nu. Of particular interest are the Alpha HPVs because of their significant contribution to oncogenesis, in which the oncogenicity varies per genotype. The HPV genome consists of three major regions. The LCR or upstream regulatory region is noncoding, and plays an important role in the regulation of viral replication and transcription of viral DNA. The other two regions, the early and late, encode eight open-reading frames (ORFs) for various proteins, including six early ORFs and two late ORFs. The early HPV genes are E1, E2, E1^E4, E5, E6, and E7. E1 and E2 are replication factors, while E1^E4, E5, E6, and E7 are accessory proteins. The late ORFs are L1 and L2, both of which encode viral capsid proteins . HPV genotypes have also been categorized into high-risk (hrHPV) or low-risk (lrHPV) groups. This classification is based on their oncogenic potential or disease association. hrHPVs are associated with cancer, while lrHPVs cause benign lesions. The hrHPV genotypes 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68 are defined by the World Health Organization (WHO) as cancer-causing types . From these hrHPV genotypes, types 16 and 18 are responsible for more than 70% of global cervical cancer cases. In general, an HPV infection is acquired during sexual intercourse, but even skin-to-skin contact may be sufficient to become infected
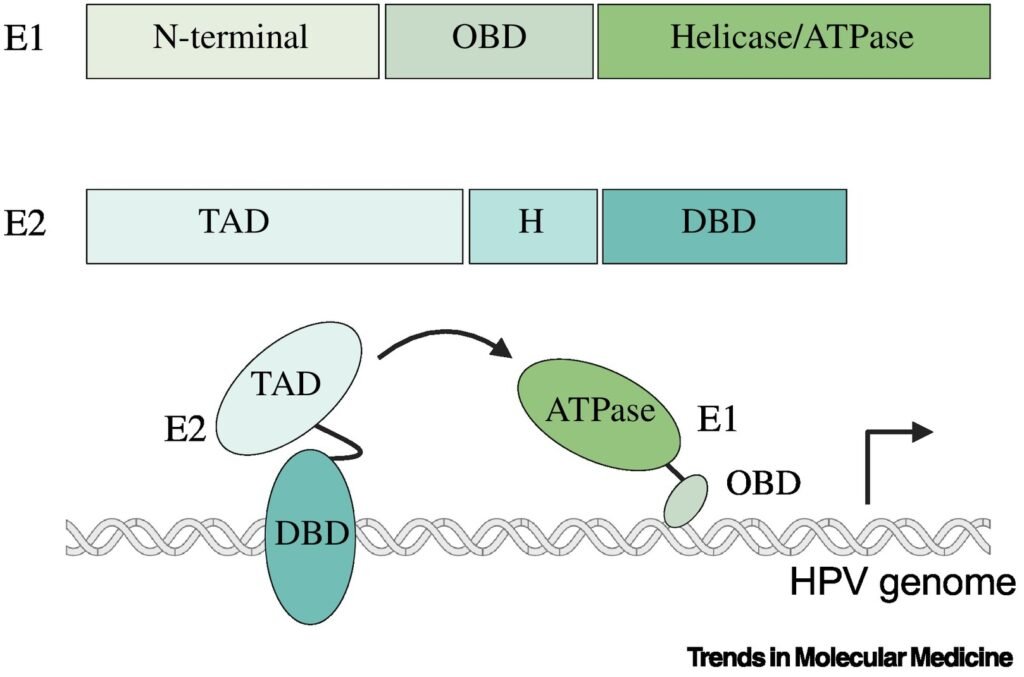
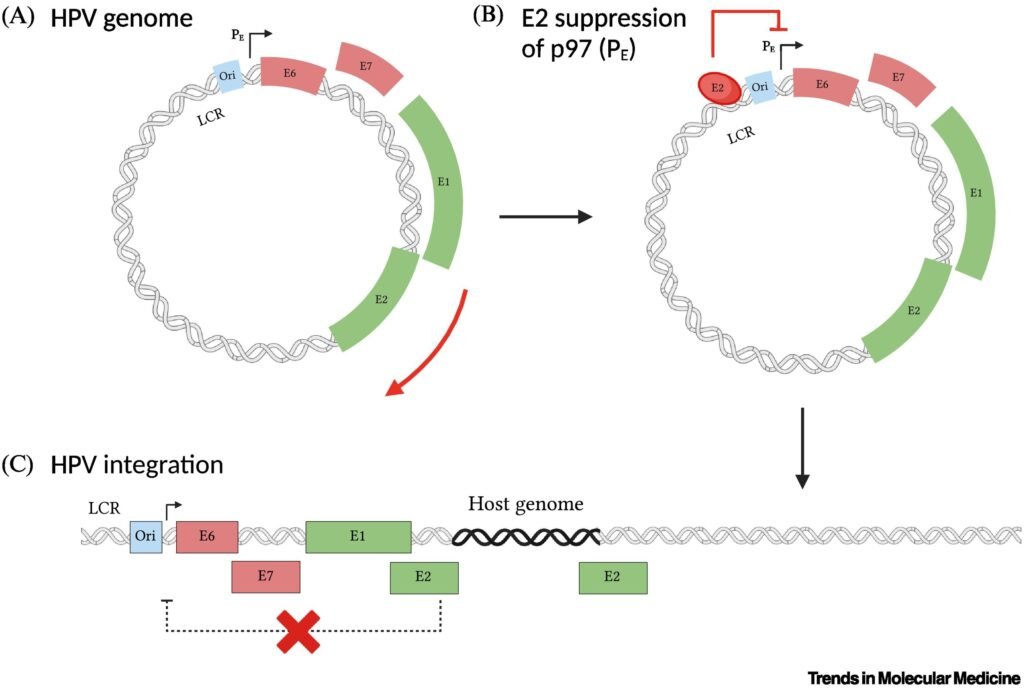
HPV DNA integration into the host genome is a complex process with advantages and drawbacks for the virus. DNA integration allows HPV to establish a persistent infection within host cells by ensuring the maintenance and replication of viral DNA along with host cellular DNA. Integration can also lead to increased expression of E6 and E7, which promotes viral replication and propagation within the stratified cervical epithelium. Likewise, integration may facilitate immune evasion mechanisms by downregulating viral antigen expression or interfering with host immune responses, thereby allowing HPV-infected and HPV-transformed cells to evade detection and clearance by the host immune system . Alternatively, loss of E2-mediated control over viral replication and gene expression can lead to dysregulated viral replication, genomic instability, and increased risk of host cell transformation. Integration therefore decreases the infectivity of HPV, as integrated viral DNA is not efficiently packaged into virions and transmitted to new host cells, limiting the spread of the virus within the host population. Moreover, HPV-induced cancer represents a dead-end for the virus, as it may ultimately lead to the death of the host and the cessation of viral transmission.
HPV DNA integration is a multifactorial process that depends on viral factors and cofactors within the host and cervicovaginal microenvironment. It is thought that one of the major cofactors of HPV integration is smoking . Smoking causes genotoxic effects in the host DNA , which together with cellular oxidative and nitrosative stresses lead to DNA damage. In women, smoking can also cause a stimulation and accumulation of mutagens in the cervicovaginal mucus, compromising the integrity of the cervical epithelium and local immunity . Additional cofactors of HPV integration might include the composition of the cervicovaginal microbiota, particularly highly diverse microbiomes with abundant pathogenic anaerobic species, estrogen levels , mental stress , and coinfection with HIV . These cofactors also increase cellular stress in the microenvironment that promotes DDRs that eventually facilitate HPV integration. Furthermore, the host genome exhibits site-specific susceptibility for HPV integration. By evaluating DNA breakpoints, it was observed that even though HPV integration occurred at random in the host, there were specific host genes that acted as hotspots and were more prone to integration . In particular, the genes FHIT, KLF5, and LINC00392 were suggested as hotspot genes based on an accumulation of integrated HPV . Similarly, another study assessed possible hotspots with evidence of HPV integration and found the gene MACROD2 as a main integration hotspot for HPV . Interestingly, MACROD2 can lead to chromosomal instability and dysregulate PARP1, an enzyme involved in cellular differentiation, proliferation, and tumor transformation .
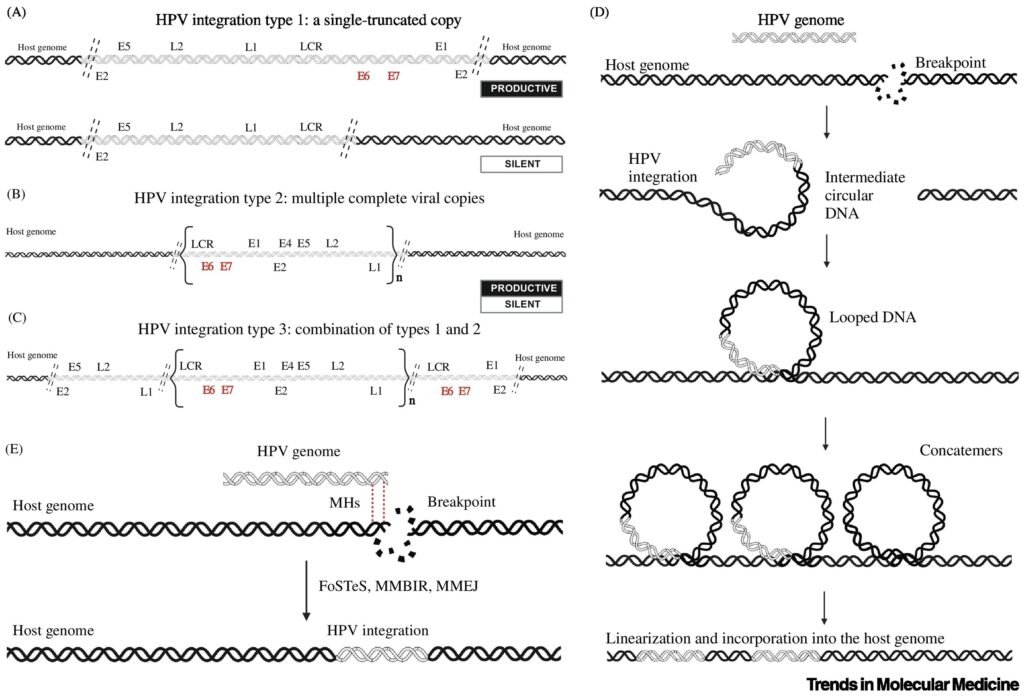
HPV integration results in host genomic instability in the chromosomes and gene alterations, particularly loss of function of tumor suppressor genes, chromatin reorganization, chromosomal rearrangements, and epigenome dysregulation . Genomic instability contributes to the progression of precancerous lesions to invasive carcinoma and facilitates the acquisition of additional genetic alterations necessary for malignant transformation . Disruption of tumor suppressor genes or activation of oncogenes through proximity to integration events can drive tumorigenesis and influence the clinical behavior of cervical cancer In addition, clonal evolution driven by HPV integration may result in the emergence of subclones with distinct phenotypic characteristics, including differences in aggressiveness, metastatic potential, and response to therapy . These overall findings imply a timeline in which HPV integration initiates a cascade of events that eventually triggers oncogenesis. Once the oncogenic pathways are triggered, cervical cancer develops.
Copyright © 2025 https://probgenbiotech.com/ All Rights Reserved.